|
|
nature communications 2022 May
- Anjun Ma,
- Gang Xin &
- Qin Ma俄亥俄州立大学医学院生物医学信息学系
注:
1, 我们之前转载了该作者的另一篇综述 :
BioIntelegence:综述: 深度学习重塑单细胞数据分析模式
2, 本文写的内容有前瞻性,且其中用到的新的专有名词较多,特添加一份翻译文档在此,并增加注释,方便同行们参考。
前言:
单细胞多组学(scMulti-omics)为免疫肿瘤学带来了变革性的见解,证明了在描述新的免疫亚群和定义抗肿瘤免疫的重要调节因子方面的成功。在这里,我们举例说明 scMulti-omics 如何用于特定的肿瘤研究,并讨论其在未来可能如何发展。
背景:
肿瘤中细胞亚群的混合被认为是耐药、转移和疾病复发的重要特征之一。肿瘤微环境 (TME) 中多种免疫细胞的存在可能会深刻影响临床结果。免疫肿瘤学的一项重大挑战是确定肿瘤中免疫细胞的异质性及其分化过程。传统的分析方法,例如流式细胞术或质谱流式细胞术,严重依赖于预先存在的知识和定义细胞类型的标记(cell-type defining markers)。混合(bulk)转录组分析稀释了一小部分免疫细胞在整个基因表达模式中的贡献。为了克服这些限制,单细胞的多组学(scMulti-omics )研究可以以更高分辨率提供不同免疫亚群的详细鉴定,并提供了解免疫细胞对肿瘤进展的贡献的机会。
注:
1, bulk RNA-seq 与 single-cell RNA-seq 相对而言,指组织或者样本中多个细胞的混合状态进行测序。
2, TME 肿瘤微环境指肿瘤细胞生长环境中存在免疫细胞等复杂的微观环境。
已经建立了 30 多种单细胞测序技术,以允许同时询问表征细胞中不同遗传和表观遗传测序信息的多种模式1。这些模式包括 DNA、基因表达、染色质可及性、染色质结构组织、组蛋白修饰、蛋白质、T/B 细胞受体和 DNA 甲基化状态(图 1a)。此外,新兴的空间转录组技术能够识别空间可变基因(在不同空间位置上具有不同表达模式的基因)、组织结构预测、细胞类型定位以及 TME 中细胞间通讯的推断2、3. 这些技术为在免疫肿瘤学研究中使用 scMulti-omics 方法提供了工具。sc 多组学数据和相关分析方法提供了对以下生物学能力的见解:
(a) 定义不同患者组中的肿瘤和免疫细胞身份,
(b) 推断不同免疫库的异质性,
(c) 了解癌细胞和免疫细胞之间的交流以及 TME 内细胞异质性的分子机制,
以及 (d) 加速在许多癌症类型中发现新的发病机制和治疗方法。
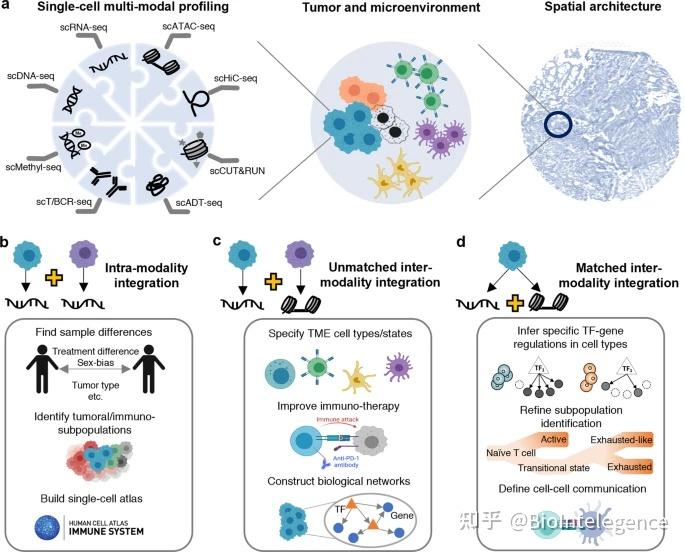
a 各种 scMulti-omics 数据类型的概述。 b-d scMulti-omics 支持免疫肿瘤学研究。
a 各种 scMulti-omics 数据类型的概述。
测序技术包括用于 DNA 序列分析的单细胞 DNA 测序 (scDNA-seq)、
用于基因表达分析的单细胞 RNA 测序 (scRNA-seq)、
用于转座酶可及染色质测序的单细胞测序分析 (scATAC-seq)染色质可及性分析、
用于染色质结构组织的单细胞高通量染色体构象测序 (scHiC-seq)、
用于组蛋白修饰分析的单细胞在靶标下切割和释放 (scCUN&RUN)、
单细胞抗体衍生标签测序 ( scADT-seq) 用于蛋白质丰度分析,
单细胞 T 细胞或 B 细胞受体测序 (scT/BCR-seq) 用于受体库多样性和克隆性分析
(指T/B 细胞受体的可变区 (V)、多变区 (D) 和连接区 (J) 基因的重组方式),
以及用于 DNA 甲基化状态分析的单细胞甲基化测序 (scMethyl-seq)。
译者注: 这些不同类型的测序技术,针对的是细胞内某一个特定的分子生物学现象,后者在下面的文字里被称为不同的模态(modality)
免疫肿瘤学中单细胞多组学的整合分析
scMulti-omics 数据的整合可分为三种类型4:(a) 模态内整合,其中从不同细胞测量相同的模态(例如基因表达)(不匹配的数据),(b) 不匹配的模态间整合从不同的细胞、样品或实验测量多种模式(例如,基因表达和染色质可及性),以及(c)匹配的模式间整合,其中从同一细胞测量多种模式。
译者注: 作者这里按照两个维度进行了分类:
1 同一个模态内 (比如只研究RNA-seq,或者只研究染色体可及性,称为模态内)
vs 跨模态/多种模态 (比如整合了RNA-seq和甲基化的分析,就是跨模态了)
2 数据集或者样本不匹配(比如分析自己实验室的RNA-seq + 甲基化。同时收集了已发表的类似细胞的数据加以比较)
vs 样本和数据集来自同一批细胞(这个实验成本最高,难度最大,结果最可靠)
在这里,我们提供了四个实验示例来证明与单个数据集的分析相比,多个单细胞 RNA 测序 (scRNA-seq) 数据的模态内整合的优势(图 1b)。第一个优势是改进细胞群识别,并能够在不同患者、治疗、时间点和物种之间进行比较分析。一项研究筛选了来自六种癌症类型的 25,149 个 CD4 + T 细胞的基因表达,并发现了一个以前被低估的肿瘤浸润性滤泡调节性 T 细胞群。该细胞亚群可有效抑制抗肿瘤 T 细胞,并与抗 PD-1 治疗的耐药性相关5. 第二个优势是能够发现广泛的不同免疫细胞类型及其基因标记。张等人。分析并整合了来自 21 种癌症类型的 316 名患者的 397,810 个 T 细胞的 scRNA-seq 数据,并描绘了 TME中 T 细胞(包括 17 个 CD8 +和 24 个 CD4 +亚群)的泛癌景观6。在每个亚型中鉴定出特定标记,例如 TNFRSF9(在调节性 T 细胞中)、ZNF683 和 CXCR6(在组织驻留记忆 T 细胞中)和 GZMK(在效应记忆细胞中)。第三个优势是整合不同的测序技术并利用它们的独特功能7。例如,使用一个Smart-Seq2(深度测序深度和高灵敏度)scRNA-seq 和10X Genomics(由于其高通量,适用于检测大细胞群)来自 CD45 +免疫细胞的 scRNA-seq 数据,Zhang 等人。将 LAMP3 +树突状细胞鉴定为源自肿瘤的重要细胞类型,迁移至肝淋巴结,并通过抗原特异性启动8塑造淋巴细胞功能。第四个优势是构建单细胞图谱的能力,例如肿瘤免疫图谱9,以便提供全面的免疫细胞纲要并检查不同免疫细胞类型中的基因表达模式。
与仅使用单个单细胞测序数据集的研究相比,非匹配的多模式整合在检测影响关键亚群的肿瘤内在和外在因素方面取得了进展。在这里,我们提供了三个示例来展示整合如何通过结合来自不同模式的独特特征来实现准确的细胞亚群预测和表征(图 1c)。非匹配的多模态整合的第一个例子是将 scRNA-seq 数据与单细胞 T 细胞受体测序 (scTCR-seq) 数据相结合,允许出现来自单个细胞(克隆型)的 T 细胞追踪子集并比较扩展、分化状态和各种克隆型之间的表型。Li等人进行的研究。发现了过渡性和功能失调的 CD8 +细胞的显着克隆共享,这将这两个 CD8 +亚群连接在一个发育途径中。这些结果提供了重要的证据来支持早期过渡 CD8 +细胞逐渐分化为功能失调的 T 细胞状态10. 在第二个例子中,科学家们使用 scRNA-seq 和scATAC-seq(转座酶可及染色质测序的单细胞测序分析)对 KMT2A 重排的急性淋巴细胞白血病以揭示年轻白血病患者的谱系可塑性显着增加11. 他们还确定了细胞毒性淋巴细胞和白血病细胞之间的免疫抑制信号通路,为分子靶向和免疫治疗方法提供了临床意义。在这个回路中,自然杀伤 (NK) T 细胞产生干扰素-γ IFNγ 以激活白血病细胞;反过来,这些白血病细胞利用抑制性分子,例如转化生长因子 β (TGF-β) 来抑制细胞毒性 T 和 NK 细胞。第三个例子展示了可以整合蛋白质丰度、基因表达和染色质可及性来识别连接基因之间癌症/免疫特异性关系的生物网络,例如顺式调节元件、TFs 和癌症相关的peak-基因联系12 , 13. 具体来说,Granja 等人。使用 Seurat 14结合在六个混合表型急性白血病样本中测量的上述三种模式13。他们观察了患者的常见恶性特征和患者特异性调控特征,并确定了 91,601 个推定的峰值基因连锁和调节白血病标志基因(例如 CD69)的关键 TF(例如 RUNX1)。
匹配的多模态集成利用 scMulti-omics 联合分析的优势,以最小的细胞和模态偏差在模态之间建立更可靠的连接(图 1d)。与非匹配的数据集成相比,匹配的多模态分析仍处于起步阶段,主要是由于费用高、实验操作严格,导致在免疫肿瘤学研究中的应用较少。首先,免疫肿瘤学中常用的联合分析技术是 CITE-seq 15,其中匹配的基因表达和蛋白质丰度是从同一细胞中量化的。Leader等人。应用 CITE-seq 分析来自 8 名非小细胞肺患者的基因表达和 81 种抗体16. 他们的研究表明,CITE-seq 允许高度准确的 CD4 +和 CD8 + T 细胞聚类和注释,而基于 scRNA 的聚类无法完全解决(例如,鉴定出富含 IFNG、GZMB、LAG3、CXCL13 和 HAVCR2 的活化 CD8+ 簇表达和 PD-1、ICOS 和 CD39 蛋白丰度增加)。其次,scRNA-seq 可以与空间转录组学联合测序,以描绘 TME 中不同免疫细胞亚群之间的通信。佩尔卡等人。分析了 371,223 个细胞的 scRNA-seq,并匹配了来自 3 个具有高 CXCL13 T 细胞程序活性的结直肠肿瘤样本中 45 个感兴趣区域的空间转录组数据17. 他们发现了空间组织的细胞间相互作用,这些相互作用有助于人类结直肠肿瘤中协调的多细胞免疫反应。具体而言,来源于T细胞的IFNγ可以诱导CXCR3配体的表达,以吸引更多活化的IFNG + 和CXCL13 + T细胞和CXCL10/CXCL11 + 骨髓细胞在人肿瘤内形成空间组织的病灶。最后,scRNA-seq 和 scATAC-seq 的联合分析可以精确定义细胞类型及其分化状态,为发现新的 TF 和表观遗传机制以及构建动态基因调控网络提供了独特的机会18。不幸的是,截至今天,尚未在公共领域发表使用这种 scMulti-omics 技术的免疫肿瘤学研究。
挑战和未来前景
在免疫肿瘤学中应用 scMulti-omics 仍然存在一些挑战。首先,批量效应消除(batch effect removal)是准确综合分析的主要障碍之一,需要保留真实信号并消除样本、条件和实验之间的差异。实验上,对来自同一细胞的多种模式进行联合分析,而不是从不同细胞中分离测序,可以大大减少批次效应;在计算上,可以通过现有的基准测试方法19、20指导选择合适的工具来处理批次效应。其次,已经开发了多种计算工具,以通用风格集成 scMulti-omics 数据21,但不是专门为免疫肿瘤学中的数据分析设计或优化的。例如,在免疫肿瘤学 scMulti-omics 数据中,降维的特征数量和 Louvain 聚类的分辨率可以量身定制比用于正常组织或细胞系来的更高。此外,公认的标记基因和特征(例如,CD4 + T 细胞的 CD3 和 CD4)可以包含在数据分析中,以自动更正细胞聚类结果。第三,目前的方法在理解细胞和不同模式之间的串扰方面能力有限。这限制了 scMulti-omics 数据在推断不同细胞类型的潜在生物网络中的应用,阐明了这些网络对特定细胞类型中外部刺激的反应13,并发现在 TME 中推动从一种细胞类型转变为另一种细胞类型的分子程序。最后,随着数据复杂性的增加(例如,一个数据集中有一千万个单元),计算效率变得更加关键,并且需要可扩展性来处理大量数据。
我们设想更多的 scMulti-omics 数据和计算工具可用于免疫肿瘤学的综合分析。随着更多数据的产生,需要系统地收集免疫肿瘤学中处理过的单细胞数据的数据库(例如,TISCH 22)。此类数据库可以为工具开发和优化方面的创新研究开辟道路,并为跨不同癌症类型、物种和治疗条件的不同方式的潜在整合提供机会。随着 scMulti-omics 数据生成的增加,深度学习将彻底改变工具开发和单细胞数据分析,因为深度学习框架在以无假设的方式从复杂数据中提取特征方面非常强大23. 例如,可以使用深度图表示学习框架(例如,图转换器)来提取和学习不同模态的适当特征,以表征细胞异质性。它具有协同识别细胞和多种模式的联合嵌入的巨大潜力,具有在同一图中包含细胞、基因、染色质峰和其他表观遗传元素的异构图模型(图 2a)。此外,在不同分析步骤高度模块化的情况下,可以使用具有可组合元素的端到端深度学习框架的设计,用户可以通过删除或添加步骤来自定义框架。通过这种方式,可以同时训练框架中的所有不同步骤,而不是按顺序进行训练。最后,需要更多的湿实验室努力来验证 scMulti-omics 数据的综合分析结果。单细胞 CRISPR 筛选是一种选择,可以一次测试有限数量的基因组和非基因组位点,以观察靶基因或表观遗传因素的扰动效应24(图 2b)
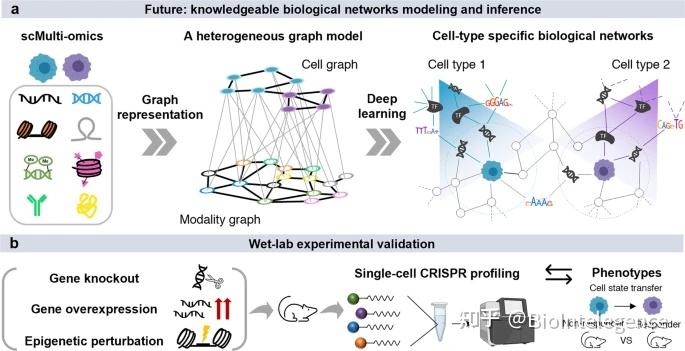
a异构图和深度学习模型可以从 scMulti-omics 数据中进行复杂的生物网络推理。b湿实验室实验验证将 scMulti-omics 预测结果与表型变化联系起来。 |
|



 发表于 2022-9-21 20:47:32
发表于 2022-9-21 20:47:32