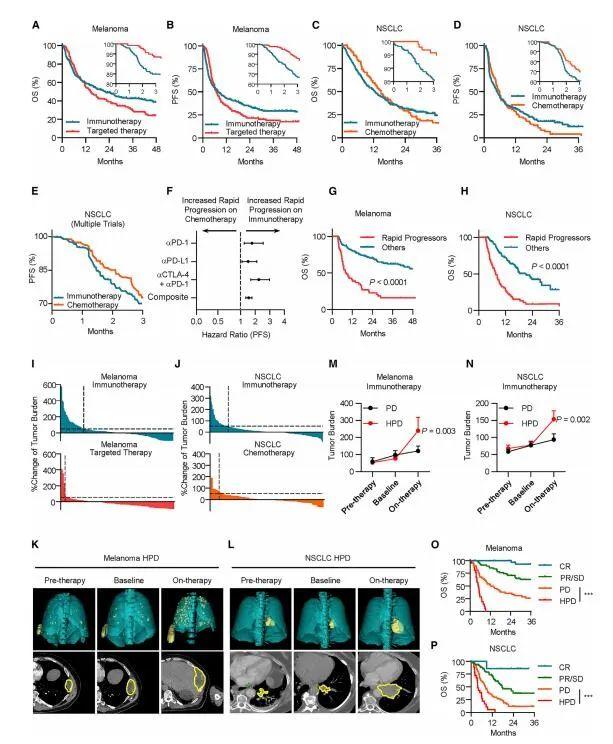
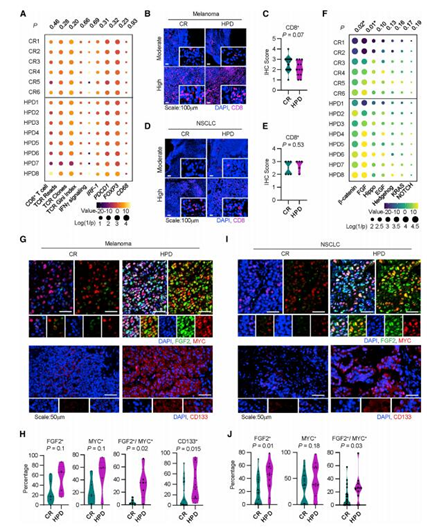
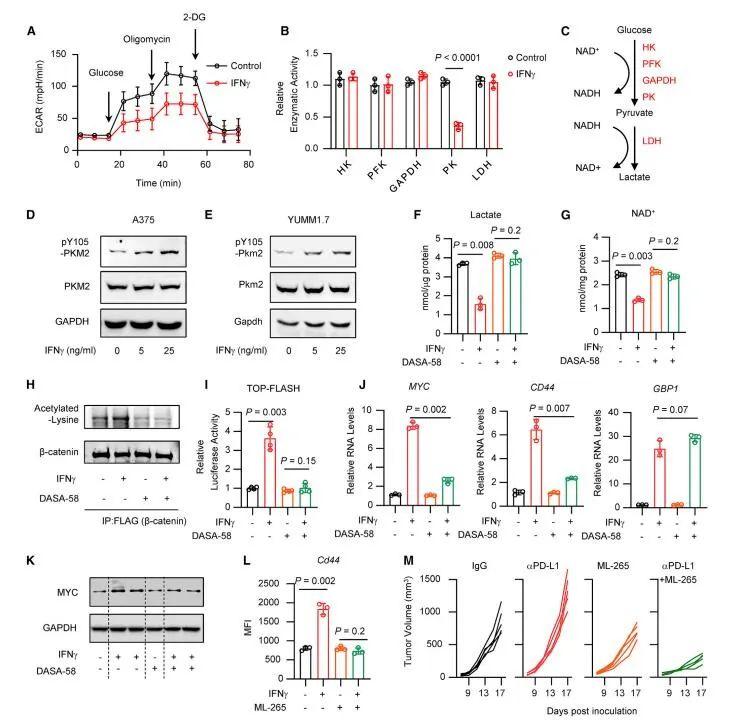
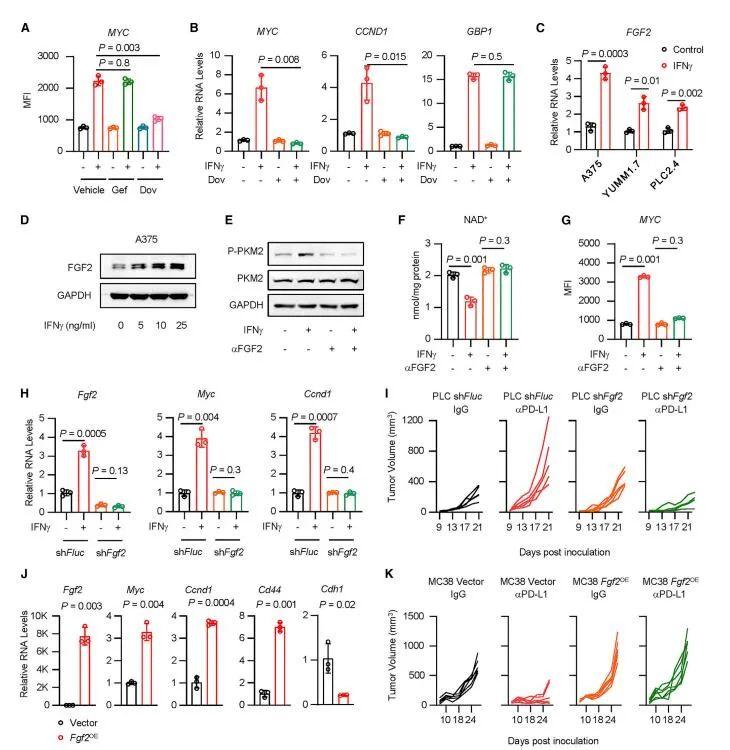
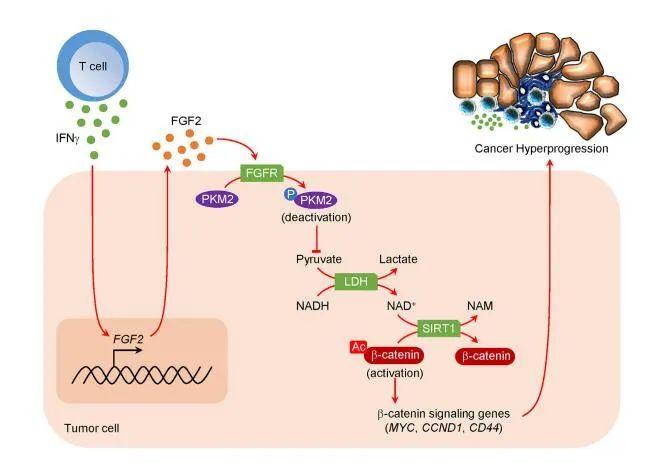
密歇根大学Weiping Zou教授课题组在国际知名期刊Cancer Cell在线发表题为“Intersection of immune and oncometabolic pathways drives cancer hyperprogression during immunotherapy”的论文。免疫检查点阻断(ICB)可以产生对癌症的持久反应。研究人员发现一部分患者在免疫治疗期间经历了反常的癌症快速进展。人们对ICB期间肿瘤如何加速其进展了解甚少。
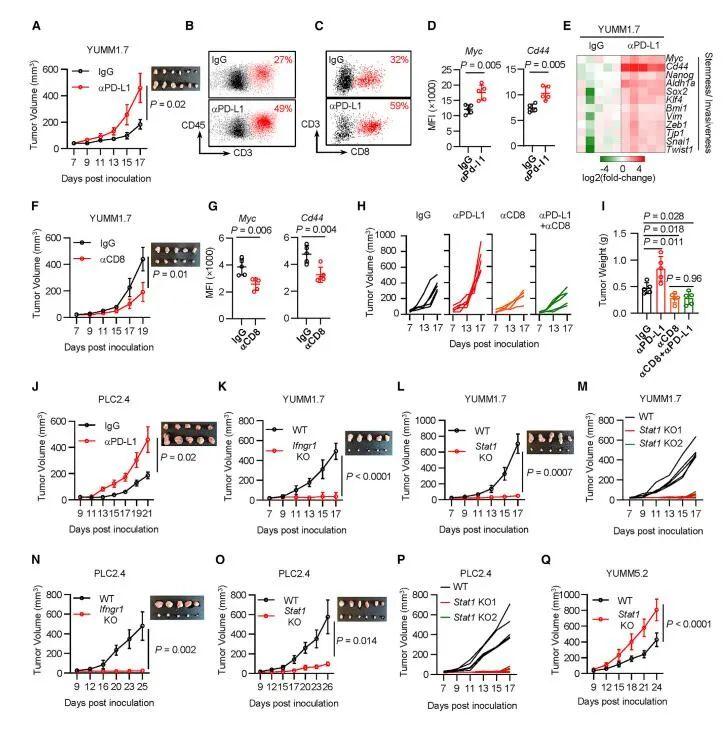
为了确定IFNγ信号通路在体内YUMM1.7肿瘤进展中的直接作用,作者汇集了3个不同的Ifng1 KO YUMM1.7克隆和3个不同Stat1 KO YUMMA.7克隆,并将这些KO克隆和WT细胞接种到WT C57BL/6小鼠中。观察到WT肿瘤在治疗过程中进展迅速,而Ifng1 KO(图3K)和Stat1 KO(见图3L)肿瘤生长缓慢。当在体内研究单个Stat1 KO YUMM1.7细胞克隆时,获得了类似的结果(图3M)。
此外,与体内WT PLC2.4肿瘤相比,合并的Ifng1 KO(图3N)或Stat1 KO(见图3O)PLC2.4肿瘤进展更慢。在单个Stat1 KO PLC2.4克隆中观察到类似的结果(图3P)。因此,IFNγ信号传导促进PLC2.4模型中的肿瘤生长。与Stat1 KO YUMM1.7肿瘤模型的结果相反,Stat1 KO YUMM5.2肿瘤的生长速度快于其WT对应物(图3Q)。因此,IFNγ信号可能在临床前模型中促进肿瘤进展。
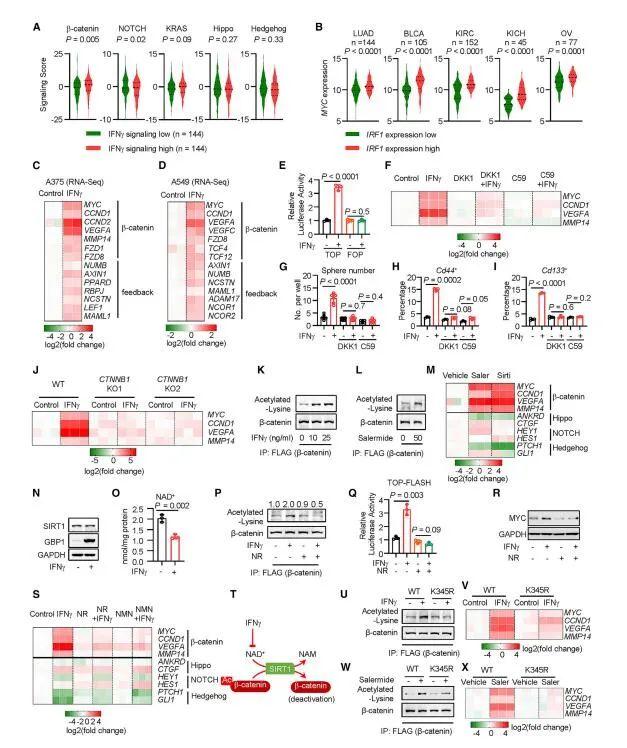
为了确定增加的基因表达是否依赖于Wnt/β-catenin信号传导,在IFNγ存在下用Wnt-β-catenin信号抑制剂DKK1和Wnt-C59培养A375和YUMM1.7细胞。这两种抑制剂降低了IFNγ介导的β-catenin信号基因的表达(图4F)、肿瘤球体的形成(图4G)和干细胞标志物Cd44和Cd133的表面表达(图4H和4I)。此外,作者建立了β-catenin KO A375细胞。IFNγ激活了WT细胞中MYC、CCND1、VEGFA和MMP14的表达,但在CTNNB1 KO细胞中没有激活(图4J)。总之,这些数据表明IFNγ激活肿瘤细胞中的β-catenin信号。
参考文献
Li G, Choi JE, Kryczek I, et al. Intersection of immune and oncometabolic pathways drives cancer hyperprogression during immunotherapy. Cancer Cell. 2023;S1535-6108(22)00594-3. doi:10.1016/j.ccell.2022.12.008



 发表于 2023-2-8 12:22:20
发表于 2023-2-8 12:22:20