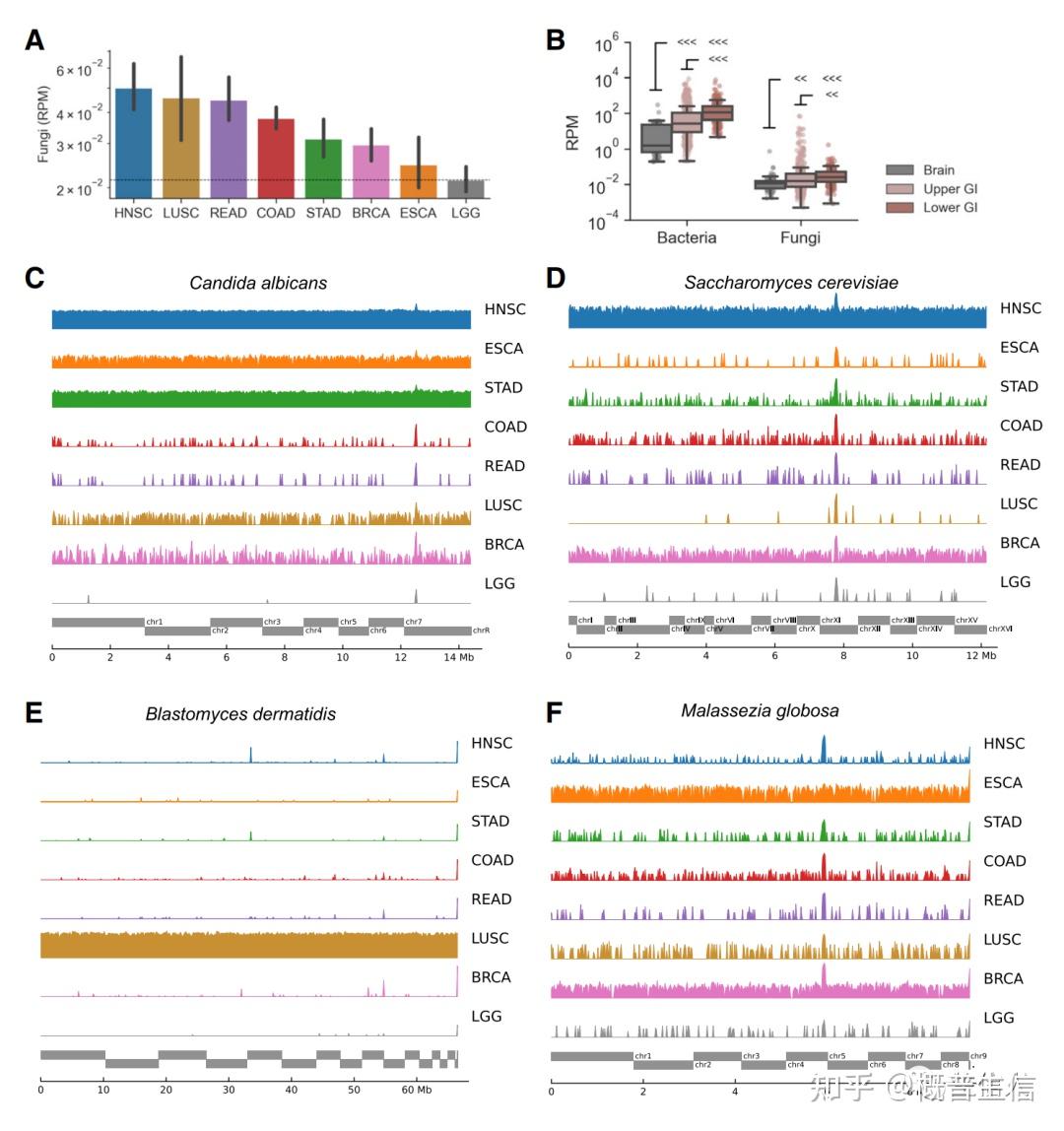
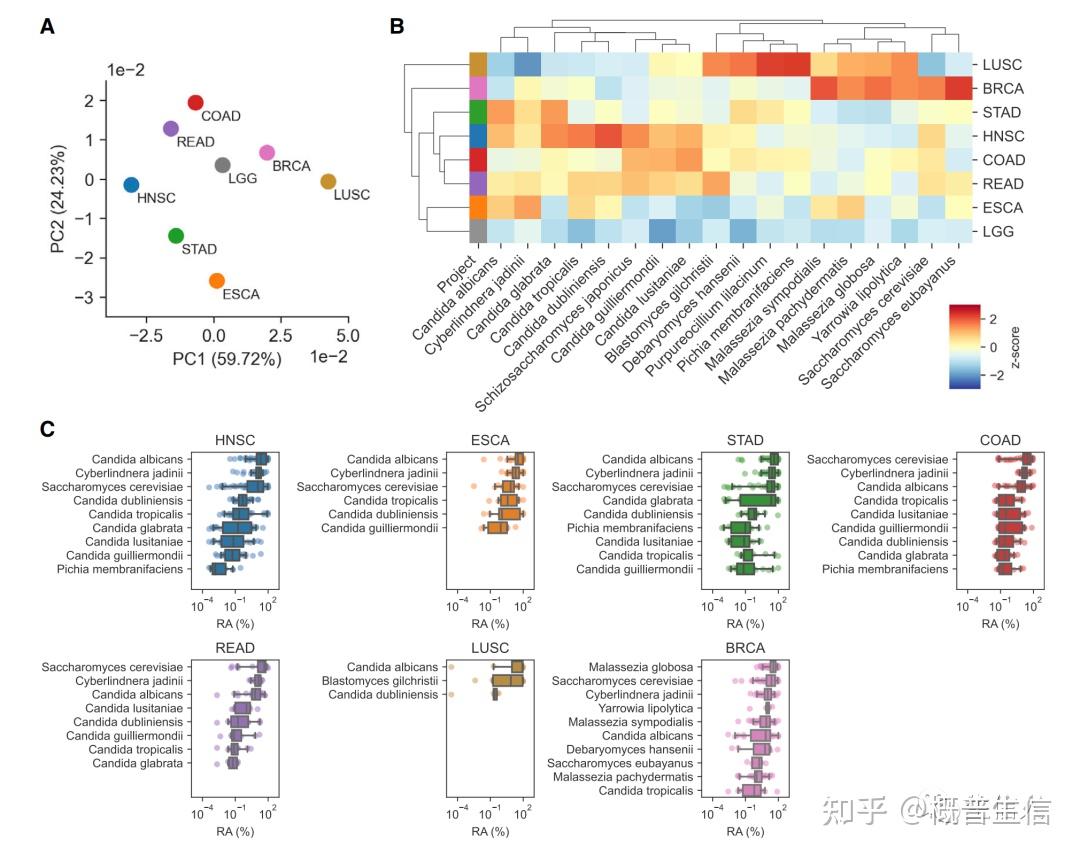
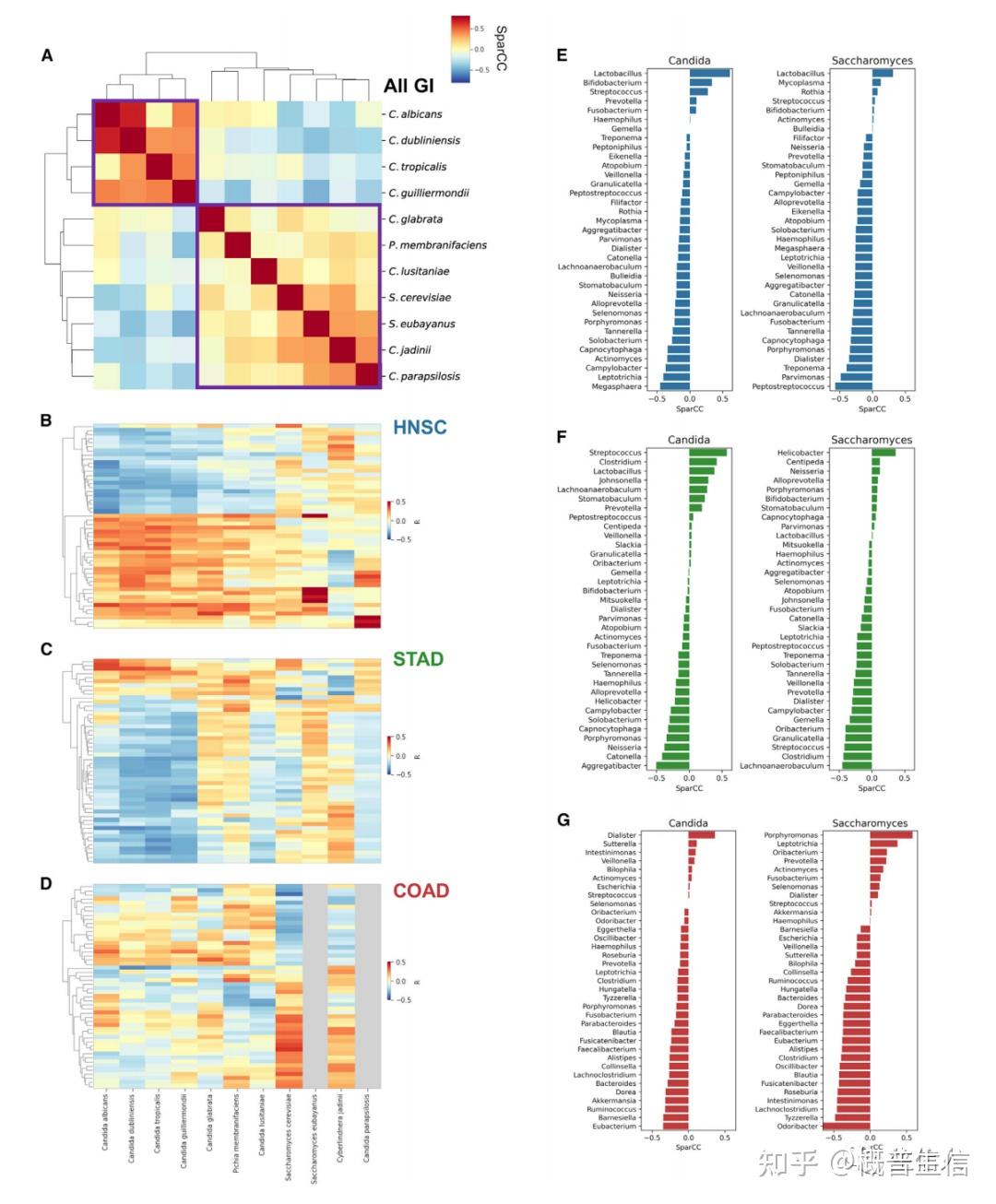
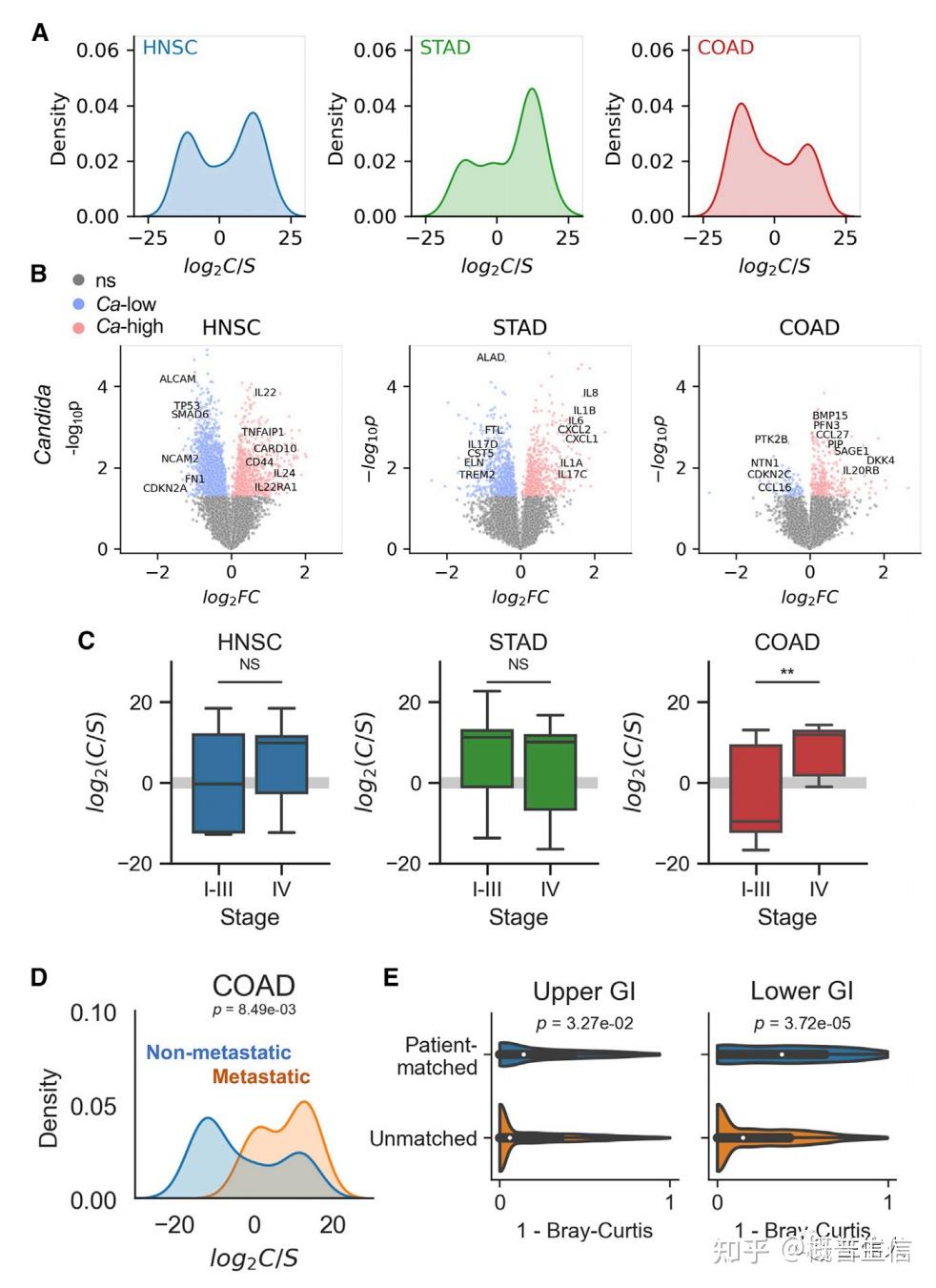
参考文献:
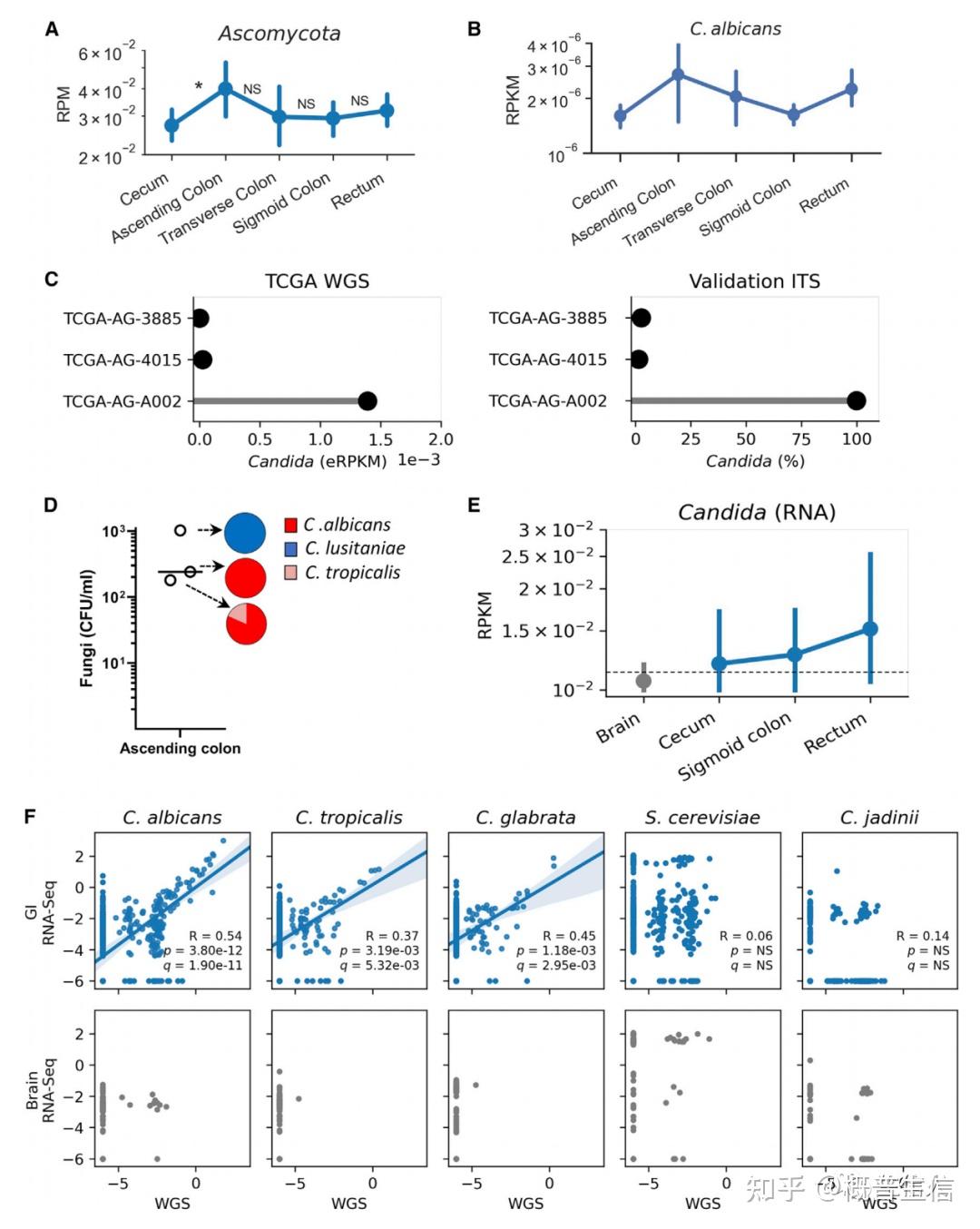
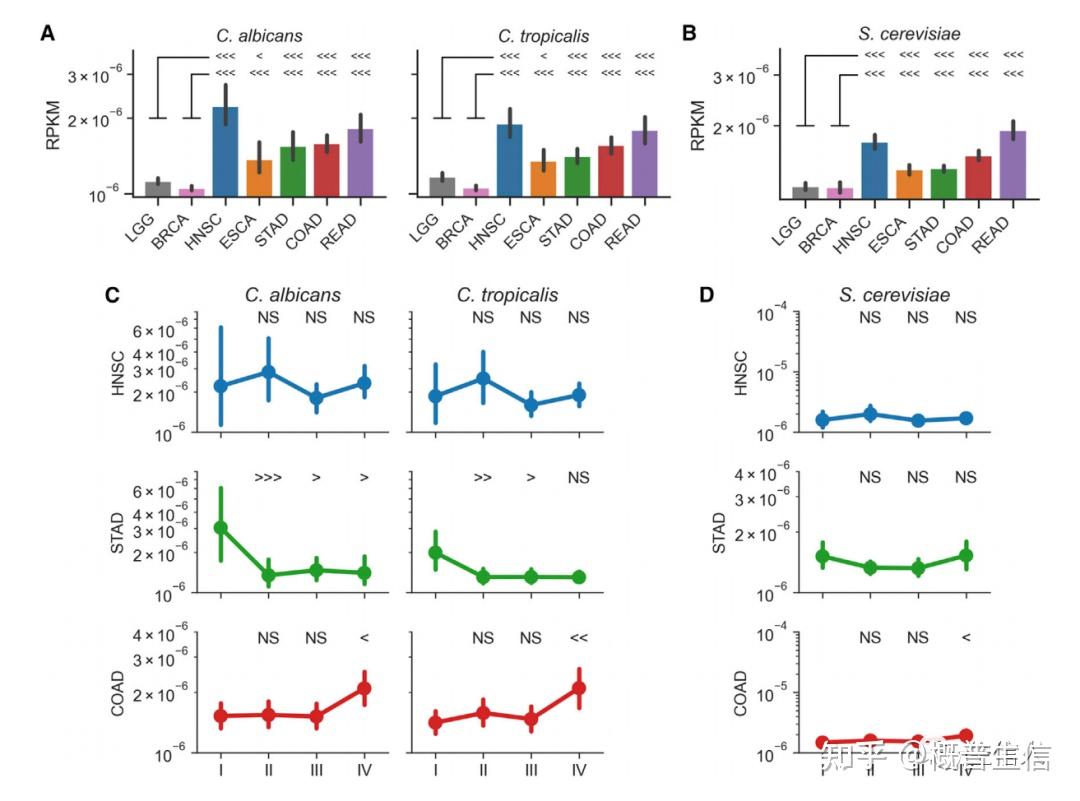
1.A pan-cancer mycobiome analysis reveals fungal involvement in gastrointestinal and lung tumors , 2022, Cell
2.BIC: a database for the transcriptional landscape of bacteria in cancer. Nucleic Acids Research, 2022
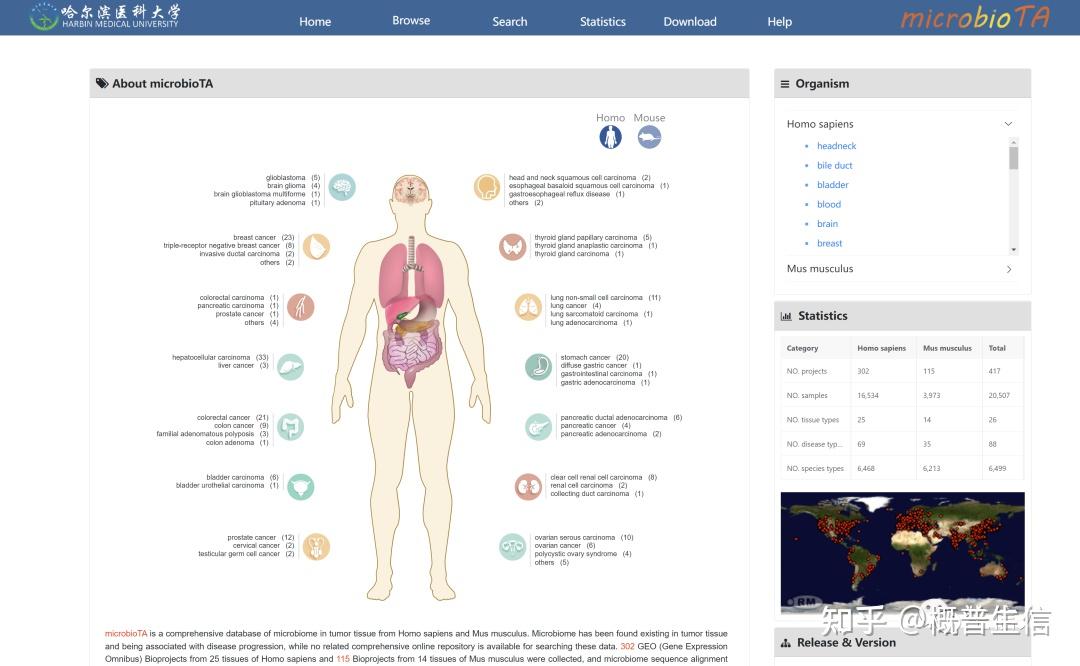
3.microbioTA: an atlas of the microbiome in multiple disease tissues of Homo sapiens and Mus musculus. Nucleic Acids Research, 2022
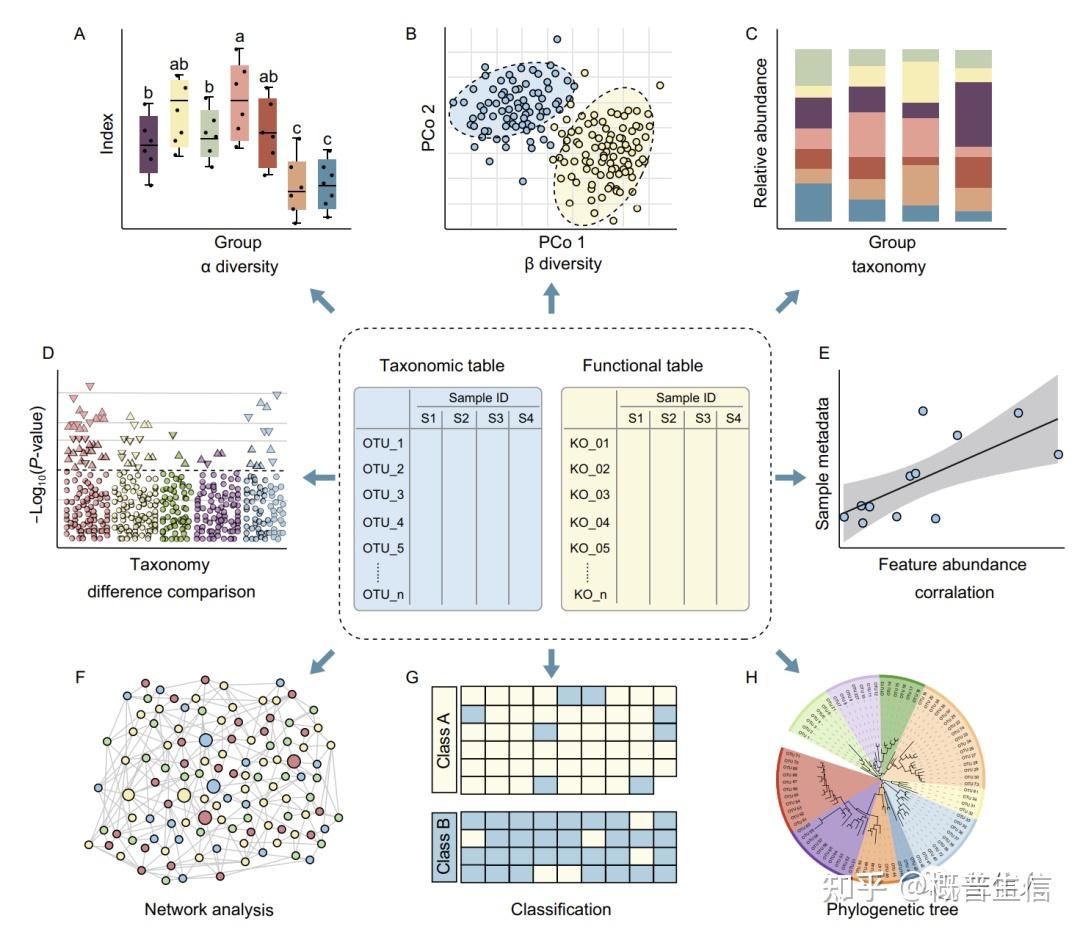
4. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein & Cell, 2020
更多热点文章推荐:



 发表于 2022-12-8 09:42:40
发表于 2022-12-8 09:42:40