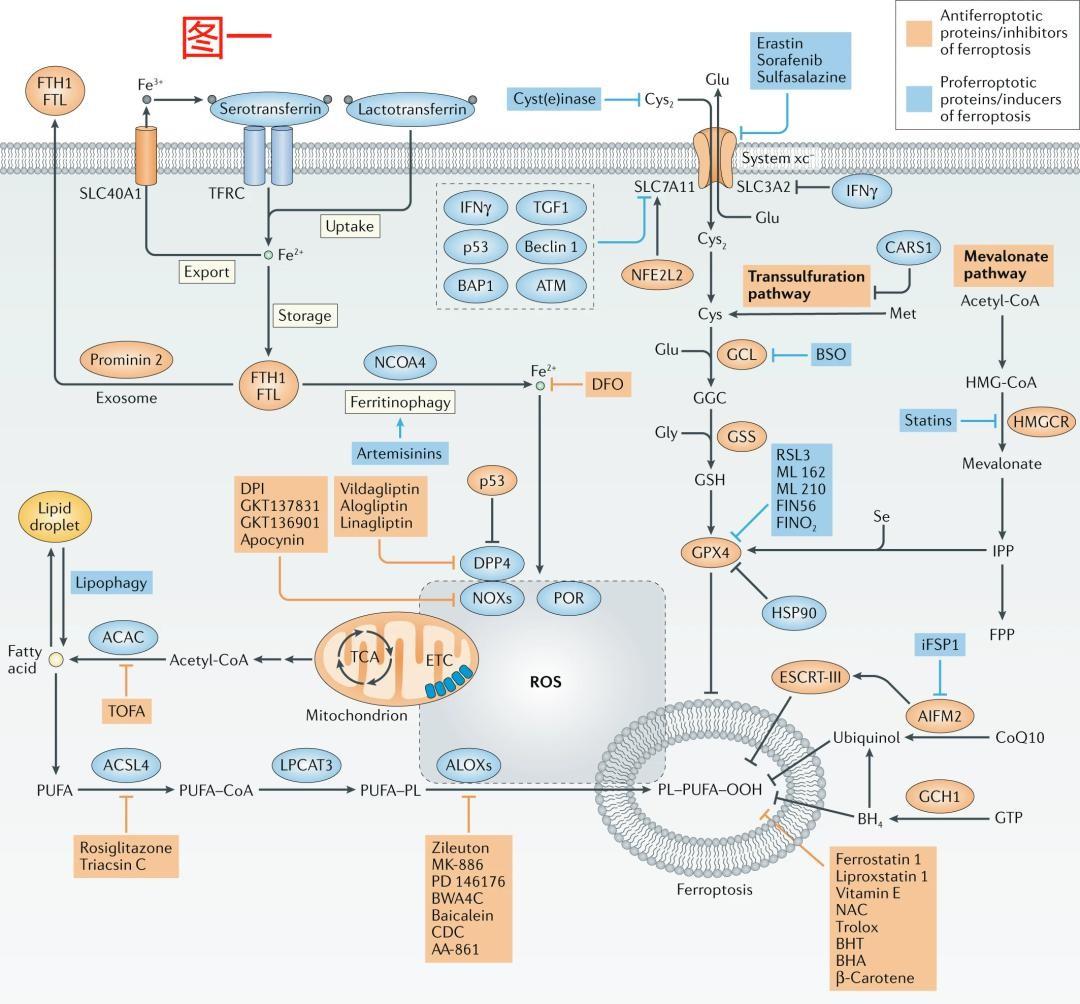
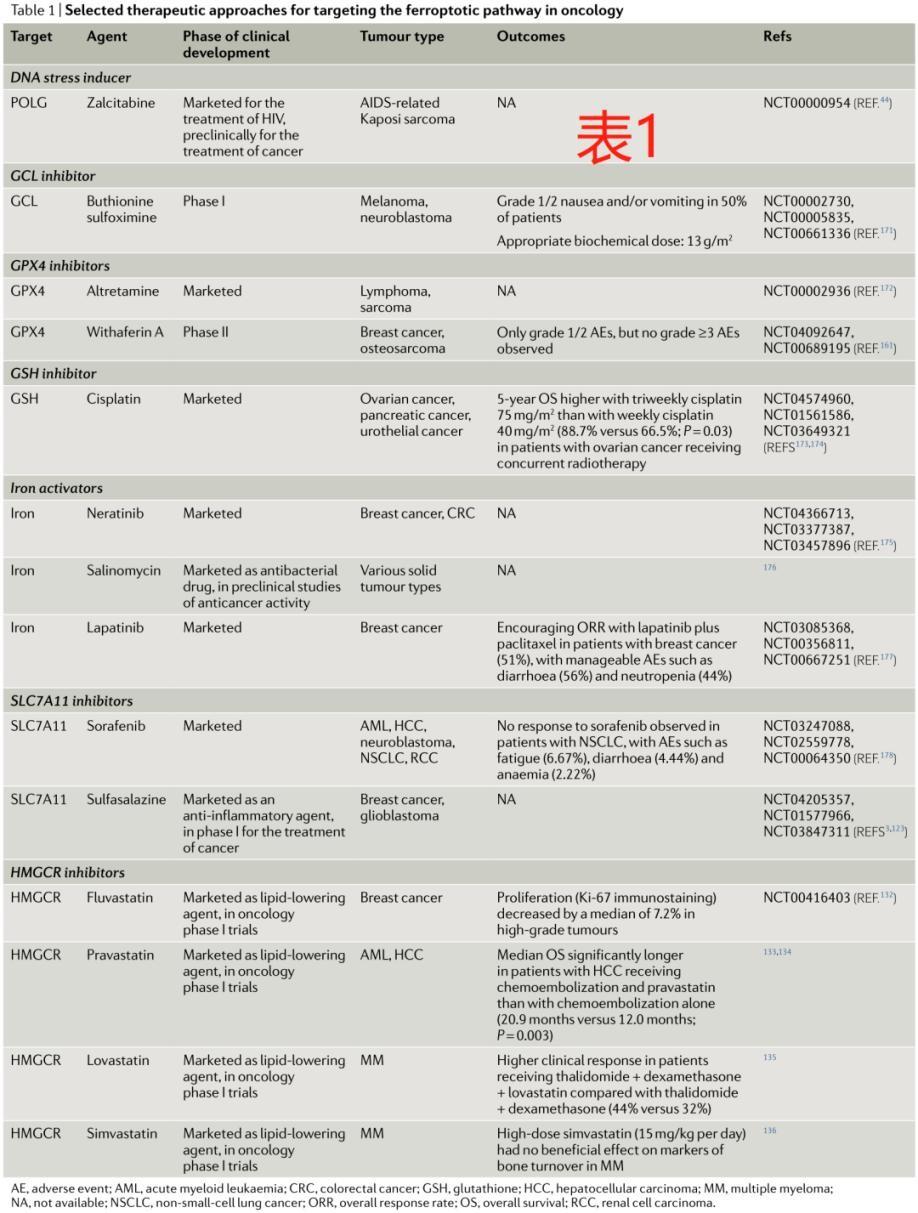
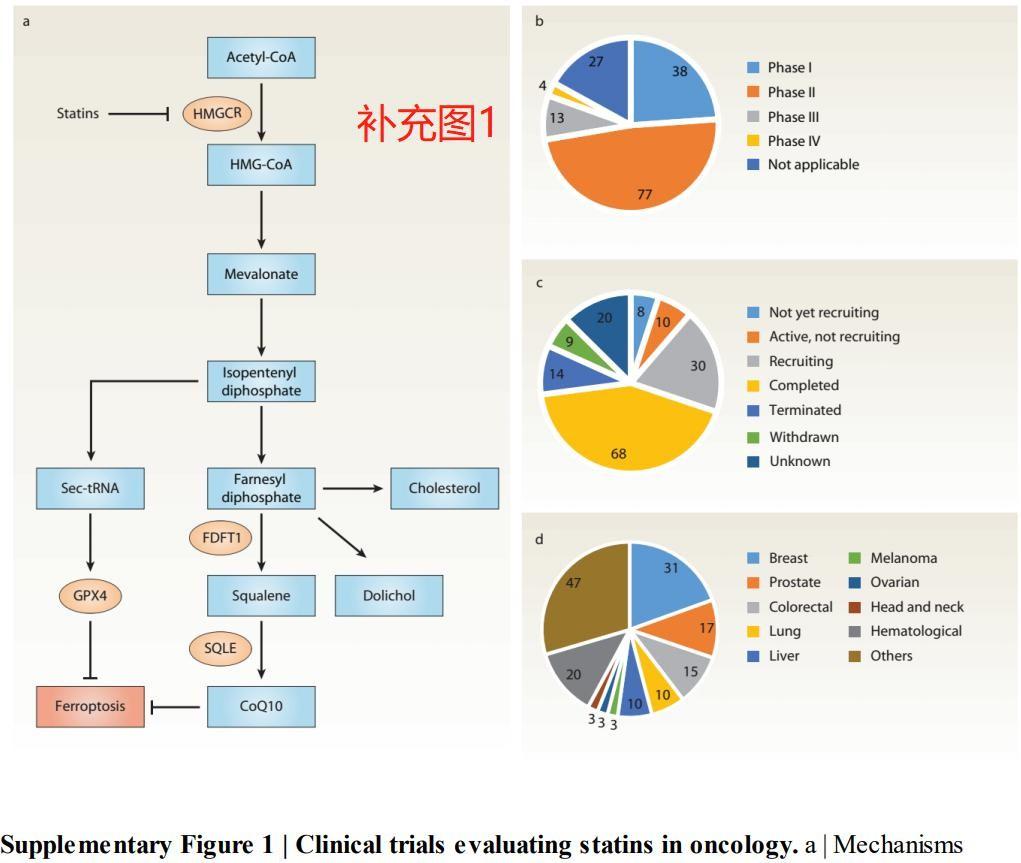
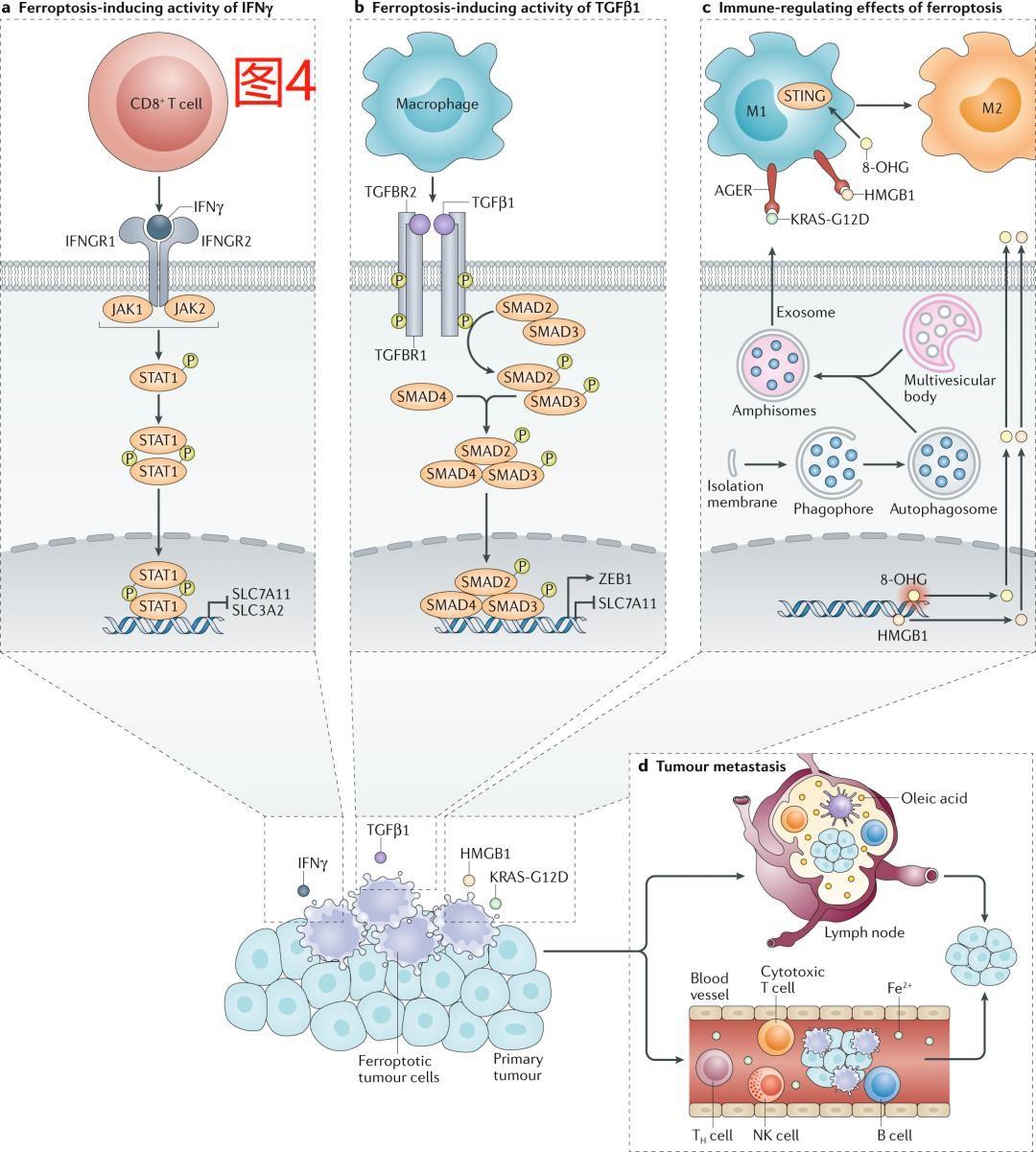
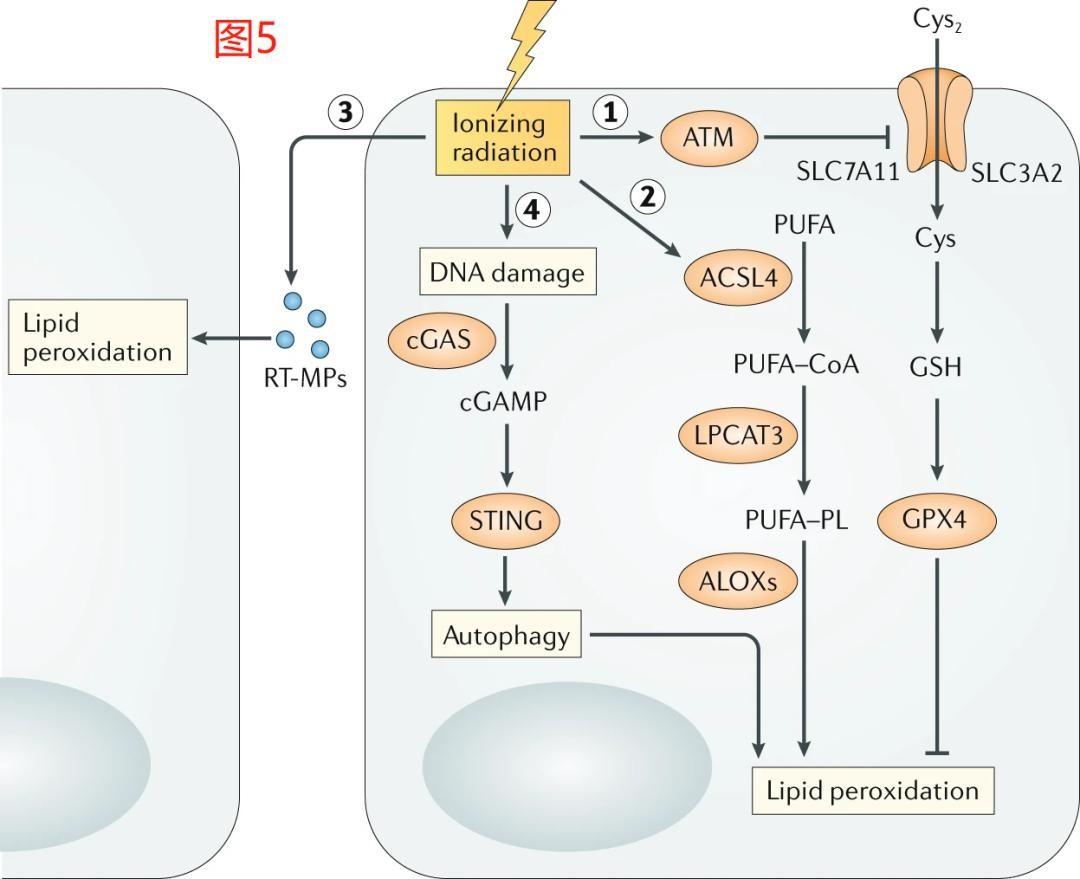
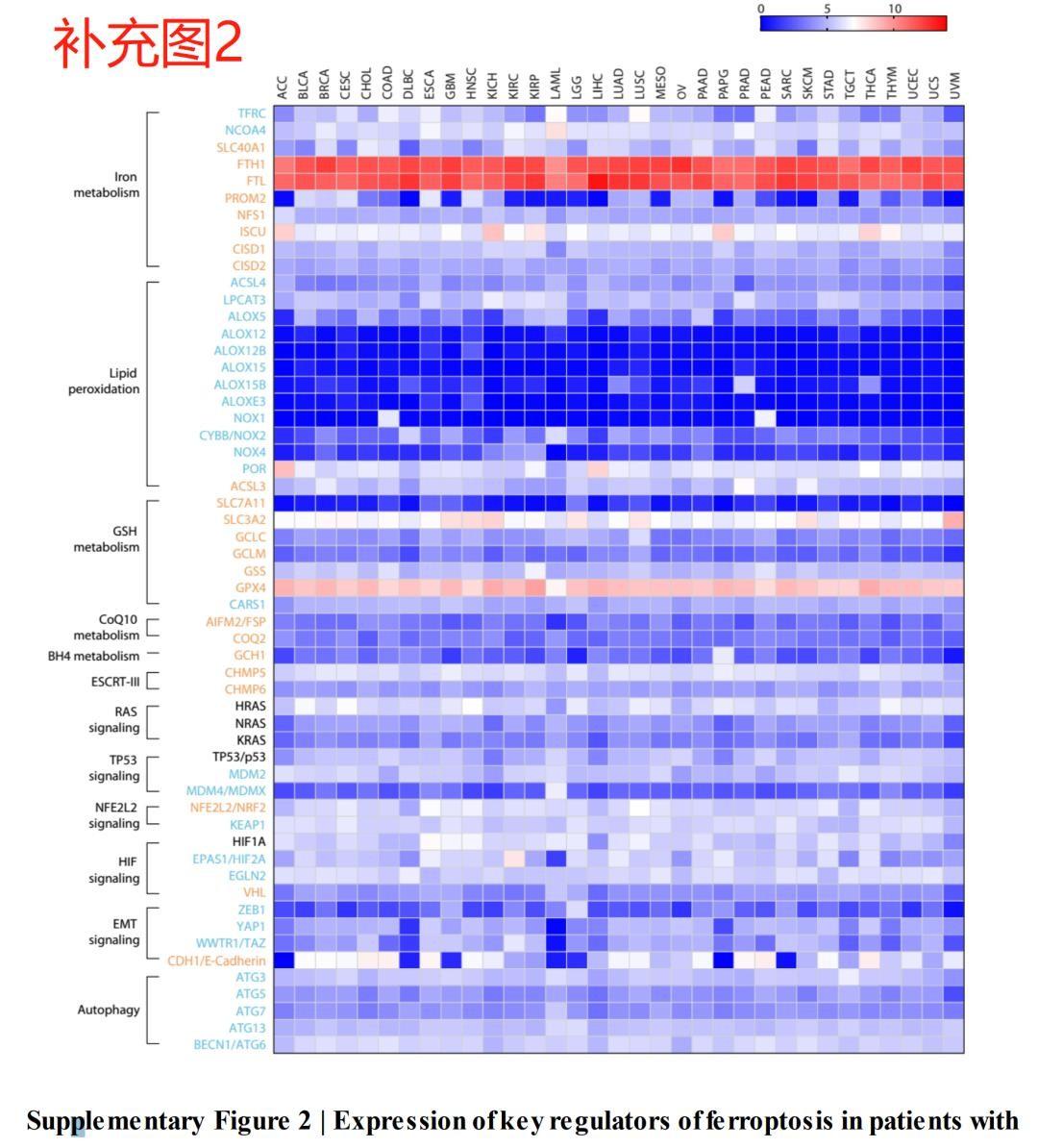
3
4
10
新手上路
使用道具 举报
2
9
19
8
18
本版积分规则 发表回复 回帖后跳转到最后一页
Archiver|手机版|小黑屋|同城医药问答网
GMT+8, 2026-3-14 04:16 , Processed in 0.147181 second(s), 20 queries .
Powered by Discuz! X3.4
© 2001-2013 Comsenz Inc.



 发表于 2022-11-30 14:50:03
发表于 2022-11-30 14:50:03