|
|
前面已跟大家分享了癌前肝脏微环境的相关内容(点击跳转原文),接下来跟大家继续分享肝脏中肿瘤发展的过程中相关内容。
细胞死亡是坏死性炎症的驱动因素
肝细胞死亡伴随着慢性肝病的疾病进展过程,如NASH、病毒性肝炎和肝硬化。主要以凋亡或坏死的形式进行。最近的发现表明,在慢性肝炎中,其他细胞死亡途径参与了肝脏微环境的形成,如铁死亡、氧死亡和细胞焦亡。后一种形式引起高度关注,但在不同形式的慢性肝炎的背景下尚未得到充分研究。值得注意的是,细胞死亡的类型会影响原发性肝癌的类型。
细胞外刺激通过激活细胞死亡受体,如胆汁淤积性肝脏中的FAS胆汁酸,激活T细胞表达的FAS配体(FASL),或通过促炎免疫细胞编码配体(例如TNF、淋巴毒素和TNFRSF12(也称为TNFRSF25))激活肿瘤坏死因子受体超家族,触发外源性凋亡。细胞内毒性条件,如高水平的ROS、DNA损伤或因再生能力耗尽而产生的复制应激,会激活内在细胞凋亡,其执行过程与外在形式相似。
第二种主要形式的细胞死亡可分为控制性(坏死)和非控制性(死亡)亚型。坏死,或控制性坏死,在几种肝病中都有描述,其特征是乙醇引起的肝损伤和胆汁淤积症或NASH的纤维化。坏死可通过多种受体(如TNFRSF1A、TNFRSF1B、FAS、TLR3和TLR4)或通过DNA传感器(如Z-DNA结合蛋白1(ZBP1;也称为DAI)和干扰素基因刺激因子(STING))被外源性因素激活。这诱导受体相互作用的丝氨酸/苏氨酸蛋白激酶3(RIPK3)的激活,其随后磷酸化混合谱系蛋白激酶样假激酶(MLKL)。
磷酸化MLKL形成多聚体孔,其整合并破坏质膜。相反,坏死是在压力条件下引起的,如物理、化学或热损伤。在酒精或药物中毒(例如,对乙酰氨基酚)或缺血再灌注损伤后的人体中可以发现这些情况,这会直接破坏细胞完整性,例如通过增加线粒体通透性和减少ATP生成。
因此,根据病因刺激,不同的细胞死亡途径有助于肝脏的炎症和细胞转化。值得注意的是,参与慢性炎症诱导的程序性细胞死亡的分子与DNA损伤修复和基因组稳定性之间的直接联系在一个肝脏胆汁淤积症的啮齿动物模型中被鉴定出。已经表明,半胱天冬酶8通过其众所周知的催化裂解功能执行慢性炎症诱导细胞死亡。同时,半胱天冬酶8作为全长蛋白质的多蛋白复合物的支架,独立于其催化结构域,以实现有效的DNA损伤修复。
因此,在慢性肝病中可以发现基因组不稳定性和细胞死亡之间的直接联系,至少在临床前模型中,这取决于半胱天冬酶8。含有半胱天冬酶8-的复合物触发γH2AX磷酸化,控制DNA完整性,因此可能防止恶性转化。抗凋亡的肝细胞下调(例如,通过表观遗传机制)半胱天冬酶8的表达,从而随着时间的推移积累更多的DNA损伤,因为由半胱天冬酶8稳定的DNA损伤修复复合物受损。
由于失去了完整的细胞膜,垂死的肝细胞释放出大量损伤相关的分子模式。与过量酒精或脂肪酸分解代谢产生的生物产物和异常代谢产物一起,这些“内源性抗原”有助于平衡免疫耐受特性的失衡。KCs的反应首先是通过开启模式识别受体信号,产生广泛的促炎细胞因子,包括IL-1、IL-6和TNF,以及趋化因子,包括CC-趋化因子配体1(CCL1)、CCL2和CCL5(参考文献22)。这导致细胞粘附分子ICAM1和VCAM1的表达增加,以及LSEC上血小板/内皮细胞粘附分子1(PECAM1)的下调,用于将单核细胞、中性粒细胞和血小板招募到应激位点,适应性免疫细胞和炎性单核细胞被招募到发炎的肝脏。
适应性免疫在慢性肝脏炎症中的实际作用因病理背景或病因而异,仍需进一步研究。已检测到CD4+和CD8+T细胞在肝脏的积聚。在NASH小鼠模型和NASH75患者中,CD4+T细胞被证明有助于高水平的IFNγ,从而加速肝脏炎症。同样,据报道,代谢活化的CD8+T细胞会加剧NASH的进展,因为CD8+T淋巴细胞的免疫耗竭可以防止小鼠模型中NASH诱导的肝损伤。有趣的是,在HBV感染的背景下,CD4+T细胞最近被确定为与肝损害相关的慢性病毒感染患者中主要产生TNF的人群。
肝脏中的肿瘤转化
肿瘤发生是正常细胞获得生存优势并逐渐积累致癌突变的过程。慢性肝损伤和炎症诱导一种独特的肝反应,称为导管反应,其特征是门静脉附近胆管周围的肝脏A6+KRT19+祖细胞样细胞过度增殖。这种代偿性再生过程有助于恢复器官结构并维持其功能。然而,当细胞在有利于基因突变积累和致癌信号通路诱导的环境中快速增殖时,增殖细胞被赋予了恶性潜能。
01肿瘤-促进炎症信号
体内研究表明,持续和未解决的肝脏炎症是致癌原。KCs分泌的促炎性TNF显示,在存在氧化应激的情况下,通过JNK信号的激活和WNT–β-连环蛋白信号的激活,触发肿瘤发生。在Mdr2(也称为Abcb4)小鼠敲除和饮食诱导或遗传诱导的肥胖模型中,TNF介导的炎症促进了HCC的形成。发现编码淋巴毒素-β、TNFSF14及其靶点CCL17和CCL20的基因高度过表达,表明淋巴毒素激活NF-κB通路。其他炎症介质包括IL-1β、IL-6、IL-11和IL-23也是促肿瘤细胞因子。其中,IL-6被认为是二乙基亚硝胺诱导的肝癌发生中最相关的候选物,也是预测病毒性肝炎向HCC转变的可靠标志物。此外,炎症环境中血管生成素2、血管内皮生长因子(VEGFs)、CXC-趋化因子配体1(CXCL1)和CXCL8的水平也有所升高,刺激血管生成,这是肝脏肿瘤发生的重要过程。
炎症诱导的持续代偿性细胞死亡和增殖加剧了氧化应激,导致肝实质细胞DNA损伤和基因突变。由活性氧或活性氮物质产生的8-氧-7,8-二氢-20-脱氧鸟苷(8-氧代-G)和8-硝基胍的致突变损伤抑制DNA修复机制中的关键酶。如果没有适当的修复机制,受损的DNA会导致基因组不稳定,并进一步提高突变频率。例如,肝癌中最常见的突变基因TP53的改变被认为是由氧化应激引起的,还通过表观遗传重塑(例如基于microRNA(miRNA)的沉默和DNA甲基化)诱导失配修复酶的失活。
编码错配修复蛋白和肿瘤抑制剂(如mutL同源物1(MLH1)和p53)的基因的高甲基化抑制了它们的表达,有利于肿瘤的发生。在氧化应激的存在下,细胞癌基因的激活和细胞-细胞融合诱导肝细胞衰老。如果没有遗传和免疫监测系统的有效消除,这些衰老细胞获得了癌前潜能。与内质网应激类似,氧化应激也可能导致炎症。在NASH诱导的HCC小鼠模型中,活化的免疫细胞充当ROS产生以及活性氮中间体的来源,并且ROS产生的增加已被证明可诱导T细胞蛋白酪氨酸磷酸酶(TCPTP;也称为PTPN2)激活STAT1和STAT3信号。STAT1转录激活通过上调CXCL9和脂质运载蛋白2的表达促进肝脏炎症。
02肝细胞可塑性与转化
了解原发性肝癌的谱系起源和细胞可塑性变得非常重要,因为具有混合HCC和CCA表型的肿瘤正在增加,并且这些肿瘤显示恶性程度升高,预后较差。最初,HCC起源于肝细胞,CCA起源于胆管细胞,但并非相反。最近的数据表明,这些假设不那么严格,肝肿瘤发生中的细胞系定型是可塑的。
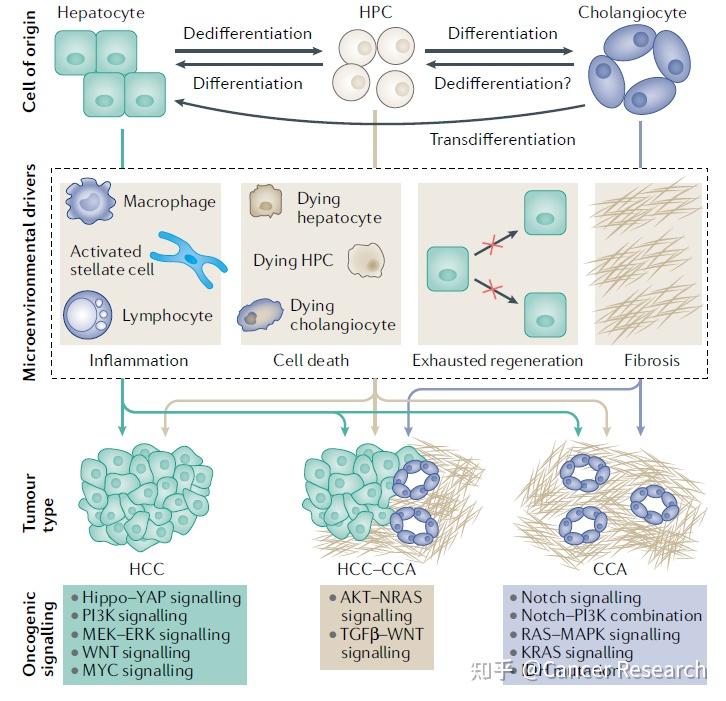
肝脏的一个独特特征是其在肝细胞损伤导致死亡时的再生能力。即使在今天,仍不清楚肝干细胞是肝脏再生的唯一来源,还是成熟的肝细胞和胆管细胞也参与了这一过程。一些研究表明,微环境可以影响这些不同细胞类型的细胞系定型,突出了肝细胞的可塑性。在硫代乙酰胺诱导的肝损伤小鼠模型中,可以表明KC衍生的Jagged1是Notch介导的肝细胞转化为胆管细胞所必需的。
其他研究还确定了Notch信号通路及其下游SOX9或RBPJ-HES1轴的激活是肝细胞可塑性的主要调节因子。另一个可能在肝细胞可塑性中起重要作用的途径是WNT–β-catenin信号传导。在3,5-二乙氧羰基-1,4-二氢吡啶的小鼠模型中饮食诱导的肝损伤,作者认为胆管细胞中WNT–β-catenin信号的激活是通过旁分泌信号将肝细胞转分化为胆管细胞的原因。
来自四氯化碳诱导的小鼠肝损伤模型和使用Hep3B细胞的体外研究的进一步结果表明,YAP1和IGFBP3在诱导肝细胞去分化中具有协同作用。在这项研究中,作者描述了通过miRNA家族Let7或剪接因子ESRP2对IGF2BP3的反馈环调节。这些研究表明,细胞起源和最终肿瘤谱系之间存在可互换的网络。事实上,一项体内研究表明,肝细胞线粒体功能障碍导致ROS产生,从而激活巨噬细胞,巨噬细胞释放TNF并导致胆管细胞JUN–JNK通路激活,从而导致CCA发展。
然而,分子谱显示,大多数人CCA表现出胆管细胞特异性模式,而其余的则具有肝干细胞的特征,这表明肝干细胞也能够产生CCA。有趣的是,肝细胞中Hippo–YAP通路的激活通过Jagged1–Notch信号传导诱导HCC的发展,该信号传导也通过KC激活。另一项研究表明,由凋亡或坏死的肝细胞诱导的微环境中的特定细胞因子谱可分别在小鼠中将转化的肝细胞驱动成HCC或CCA谱系。该谱系受转录因子TBX3和PRDM5的表观遗传调节影响。值得注意的是,肝细胞癌患者在接受经动脉化疗栓塞(一种导致肝脏大量细胞死亡的方法)后,在推测的肝细胞癌病灶周围显示出胆管细胞特征增加。
最近的一项研究分析了大量合并或混合HCC-CCA患者的队列,表明这种类型的肿瘤具有单克隆起源,甚至CCA样区域也可能来源于同一肿瘤内的HCC区域。总之,这些结果强调了肝癌可塑性的关键作用以及微环境对肝癌亚型发展的影响,影响临床治疗反应和患者生存。
肝癌代谢景观
无论病因如何,在肝脏肿瘤中经常观察到葡萄糖、脂质和核苷酸代谢的变化。肿瘤细胞倾向于糖酵解(其燃烧葡萄糖的效率比有氧呼吸低15倍),而不是氧化磷酸化,以满足其高增殖的能量和合成代谢需求。糖酵解产生的累积代谢产物可被引导至己糖胺生物合成途径、磷酸戊糖途径、三羧酸循环、脂质合成和氨基酸合成途径,所有这些途径在肝癌中通常失调。葡萄糖和核苷酸的代谢已在其他地方进行了综述;因此,在这里,我们关注肝脏肿瘤微环境中脂质的代谢变化和免疫细胞的代谢重编程。
代谢重编程机制在NASH和ASH诱导的肝癌中至关重要,其中代谢底物的改变一方面促进肝细胞的增殖能力,另一方面改变免疫室的活性。这些机制包括不平衡的脂质摄入和脂肪酸从外周脂肪组织中动员,从而导致高水平的循环脂肪酸和葡萄糖,诱导癌细胞适应葡萄糖和脂质分解代谢是主要的能量来源。这种代谢重编程不仅赋予肝癌细胞在实质内增殖和扩散的强大能力,还产生对化疗药物的耐药性。
因此,靶向参与关键代谢途径的特定酶可能使癌细胞容易受到治疗。例如,抑制脂肪生成的硬脂酰辅酶A去饱和酶(SCD)已被证明可通过内质网应激增加使体内HCC肿瘤对索拉非尼治疗敏感。与此相一致,肝脏X受体-α(LXRα;也称为NRIH3)的药理学激活与RAF抑制相结合,最近已表明可诱导与SCD活性降低和饱和脂肪酸积累相关的癌细胞脂质毒性。类似地,在高热量诱导的体内HCC模型中,给予乙酰辅酶a羧化酶(ACC)的肝脏特异性合成抑制剂(另一种脂肪生成限速酶)可以单独或联合索拉非尼减缓HCC的发展。有趣的是,异柠檬酸脱氢酶1(IDH1)或IDH2基因的改变约占CCA中确定的遗传倾向的20%。作为三羧酸循环中的一种关键酶,IDH1和IDH2的突变可导致肿瘤代谢产物2-羟基戊二酸(2HG)的积累,通过阻断肝细胞核因子4-α(HNF4α),消除体外原代小鼠成肝细胞的分化。达沙替尼在携带IDH1或IDH2突变的CCA中抑制SRC激酶信号传导,在临床前模型中显示出有希望的治疗潜力。
在代谢改变促进致癌转化的背景下,值得一提的是,男性肝癌的发病率高于女性。最近一项研究提出了一个有趣的概念,表明在肥胖的情况下,睾酮抑制白色脂肪组织中脂联素的产生;在化学诱导的肝癌小鼠模型中,这导致雄性小鼠中AMPK信号的表达降低,肿瘤进展增加。尽管性激素似乎在这一过程中起着关键作用,但形成肝癌性别差异基础的机制仍不清楚。
代谢环境也可以塑造肝脏的免疫反应,使肿瘤细胞逃脱免疫监视机制。在喂食缺乏蛋氨酸胆碱的饮食的小鼠中,亚油酸介导的脂肪毒性导致肝CD4+T细胞选择性丢失,并促进肝癌发生。此外,免疫细胞经历代谢重编程,这是导致其功能改变的原因。HCC微环境中的巨噬细胞已显示获得M2极化;这种表型与脂肪酸氧化能力增加和体外肿瘤细胞迁移有关。T细胞代谢的变化也影响其定向和抗肿瘤活性;Treg细胞在HCC微环境中的扩张与其利用能量底物(如脂质和葡萄糖)的能力增加相关。
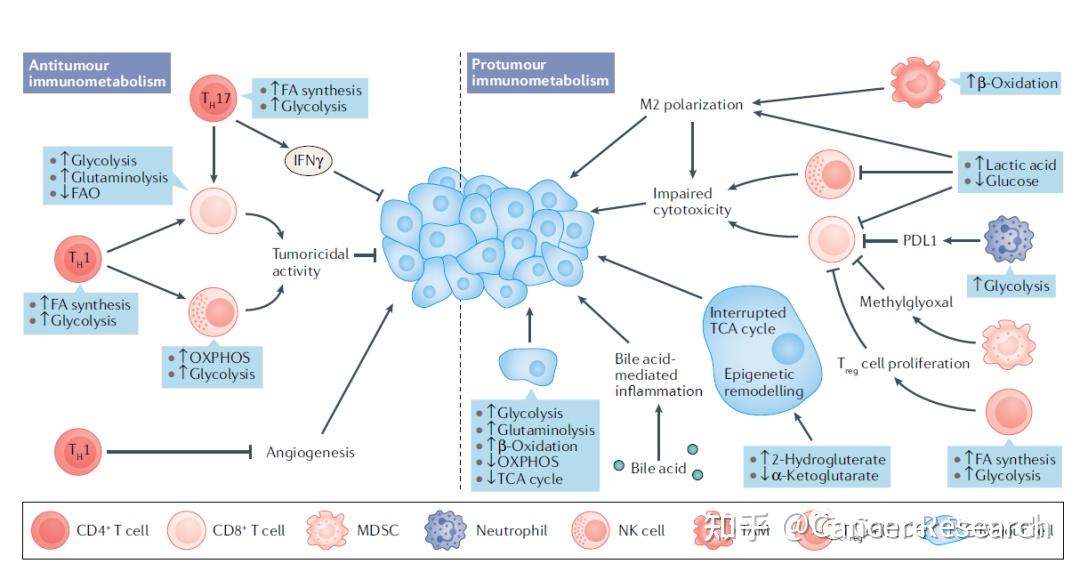
促炎性免疫细胞在肿瘤微环境中也显示出有氧糖酵解的速率增加。在这个方向上,在HCC患者中检测到丙酮酸激酶M2(PKM2)的过度表达,这与CD8+T细胞、Treg细胞和M2巨噬细胞的不良预后和免疫抑制极化有关,从而维持HCC的进展。类似地,最近研究表明,肝周单核细胞在人HCC中显示出糖酵解活性增加,这种代谢刺激诱导这些细胞中PD-L1的表达,从而抑制T淋巴细胞对肿瘤细胞的细胞毒性活性。
这篇高分综述就跟大家分享完啦~感谢大家的支持!
您的关注是对小编最大的鼓励~ 微信搜索:tumor__research,关注获取更多肿瘤研究进展! |
|



 发表于 2022-12-30 16:43:53
发表于 2022-12-30 16:43:53